AkaiKKRのインストール
AkaiKKR(machikaneyama)は、東京大学物性研の赤井 久純先生らによって開発されている第一原理計算パッケージです。 全電子法の1種である Korringa-Kohn-Rostoker (KKR) 法を採用しており coherent potential approximation (CPA) を組み合わせることにより化学的な不規則(不純物、不規則合金、固溶体)・磁気的な不規則(キュリー温度における強磁性体)などのバンド構造を計算することが可能です。
この文章ではAkaiKKR(machikaneyama)(September 10, 2020)を Ubuntu 20.04 にインストールする方法を書きます。 Virtual Box 上で、クリーンインストールをした Ubuntu 20.04 を用いて動作することを確認しました。 以降、グレーの背景で示された部分は、端末に入力するコマンドを表しています。 また、ベージュの背景は、テキストエディタなどで開いたファイルの中身を表しています。
グレーの小さい文字で書かれた部分は、本質的でないメモです。 基本的には読み飛ばして大丈夫です。
前準備
まず、なにはともあれ諸々のアップデートをしておきます。ターミナルに以下を打ち込んで、アップデートと再起動をしておきます。
sudo apt update && sudo apt -y dist-upgrade && sudo apt -y autoremove && sudo rebootAkaiKKR をインストールするために必要なパッケージを導入します。 具体的には fortran コンパイラと make と gnuplot の3つです。
sudo apt -y install gfortran build-essential gnuplot
fortran コンパイラは、有料の Intel fortran compiler (ifort) と無料の gfortran の2つの選択肢がありますが、今回は gfortran を利用します。
大学の研究者が一番安く購入できる ifort は、XLSOFTのページで確認すると税込み¥84,700の「INT7272 インテル Parallel Studio XE 2020 Composer Edition for Fortran Linux アカデミック 日本語版」であろうと思います。
赤井先生は ifort で開発をされているはずなので、予算に余裕があるなら ifort を選ぶメリットはあるかもしれません。
build-essential を指定すると他の色々なパッケージとともに make が導入されます。
別に make だけでも良いのだとは思いますが、他のDFTパッケージをインストールするならばどうせ必要になるので。
gnuplot は有名なグラフ描画アプリです。AkaiKKR パッケージの中で状態密度やバンド分散を描画するための実行ファイルである gpd や spc から呼ばれるのでインストールしておきます。
AkaiKKRのメインプログラムのインストール
AkaiKKR(machikaneyama)のソースコードの入手をします。 初めての利用の場合は登録フォームに必要事項を記入しアカウントの作成をする必要があります。 しばらくすると、登録したメールアドレスにパスワードが送られてくるのでダウンロードページにログインします。 基本的には最新版をダウンロードすればよいと思います。この文章の執筆時点では September 10, 2020 が最新版です。 ダウンロードしたアーカイブは(別にどこでも構いませんが) ~/Downloads/AkaiKKR/20200910/cpa2002v010.tgz に置いてください。
AkaiKKR のバージョンは、公開された日付で区別されています。 マイナーアップデートのように見えて挙動が大きく変わることもあるので、アップデートするときには古いバージョンを残しておく方が良いです。この文章でも日付のバージョンごとに異なるディレクトリにインストールする前提で話を進めます。
次に AkaiKKR をインストールするディレクトリを用意します。 今回は September 10, 2020 なので ~/kkr/20200910/cpa2002v010/ にインストールすることにします。
mkdir ~/kkr
mkdir ~/kkr/20200910
cd ~/kkr/20200910
tar xzvf ~/Downloads/AkaiKKR/20200910/cpa2002v010.tgz
cd cpa2002v010/AkaiKKR をコンパイルするための設定が書かれている makefile の編集を行います。 編集には使い慣れているテキストエディタを使ってください。emacs でも vim でも。 下の例では gedit を使っています。
gedit makefilemakefile の13行目から17行目にかけて fortran コンパイラーに Intel fortran Compiler を使うか gfortran を使うのかを指定する箇所があります。 今回は gfortran を使うので ifort が指定されている15行目をコメントアウト(行頭に#を追加)し、代わりに16行目の#を削除し、上書き保存します。
#######################################
# specify the fortran compiler
#fort = ifort
fort = gfortran
#######################################ターミナルに戻って make とタイプすると AkaiKKR のメインプログラムのコンパイルが始まります。 コンパイルに成功すると specx という名前の実行ファイルが生成されるはずです。
makeこの文章の執筆時点では gfortran でコンパイルすると大量の warning 出力されてびっくりしますが、生成された実行ファイルには問題はないようです。
なお、何らかの理由で AkaiKKR のプログラムを編集したい場合は、ここで編集しておきます。
興味がない場合は、次のテスト計算(go計算)へ進んで大丈夫です。
ここでは、一例としてAkaiKKRのk点メッシュ
を参考にして、k点の分割メッシュを出力できるようにします。
まず該当するソースコードを編集します。今回の場合は source/bzmesh.f です。
gedit source/bzmesh.f
ファイルの一番最後が end で終わっているので、その前の行に write(*,'(3x,3(a,i3))')'nfa=',nfa,' nfb=',nfb,' nfc=',nfc という行を追加します。
以下は編集前の最後の3行です。
80 continue
deallocate(iwt)
endこれに1行追加して、以下のようになります。
80 continue
deallocate(iwt)
write(*,'(3x,3(a,i3))')'nfa=',nfa,' nfb=',nfb,' nfc=',nfc
endソースコードの編集が終わったら再び make を行います。
makeこの make コマンドは、前回 make を実行して以降、編集したソースコードだけをコンパイルして実行ファイルを作ってくれます。
テスト計算(go計算)
in/ ディレクトリに色々な物質の入力ファイルのサンプルがあります。 ここでは鉄の計算を行ってみましょう。 テキストエディタで in/fe を開いてみると以下のようになっているはずです。
gedit in/fec----------------------Fe------------------------------------
go data/fe
c------------------------------------------------------------
c brvtyp a c/a b/a alpha beta gamma
bcc 5.27 , , , , , ,
c------------------------------------------------------------
c edelt ewidth reltyp sdftyp magtyp record
0.001 1.0 nrl mjwasa mag 2nd
c------------------------------------------------------------
c outtyp bzqlty maxitr pmix
update 8 100 0.035
c------------------------------------------------------------
c ntyp
1
c------------------------------------------------------------
c type ncmp rmt field mxl anclr conc
Fe 1 0 0.0 2 26 100
c------------------------------------------------------------
c natm
1
c------------------------------------------------------------
c atmicx(in the unit of a) atmtyp
0 0 0 Fe
c
c------ The following types of inputs and their combination
c are also allowed.
c------ In those cases a, b, b mean primitive unit vectors
c and x, y, z mean conventionnal cell vectors along
c x, y, and z axses. Bare numbers indicate cartesian
c coordinate in the unit of lattice constant a.
c 1/2 1/2 1/2 Fe
c 0a 0b 0c Fe
c 0x 0y 0z Fe
c 1/2a 1/2b 1/2c Fe
c------------------------------------------------------------
c "end" means the end of input data
c "end" is not necessary unless other data used only
c for "spc" exist below.
c----------------------Fe------------------------------------
#end
# The following data are used in the 'spc' mode.
# number of k-point along symmetry line
300
# For bcc bravais lattice, the dispersion relation along
# G-H-N-P-G-N symmetry points (below) is usually calculated.
0 0 0
0 1 0
1/2 1/2 0
1/2 1/2 1/2
0 0 0
1/2 1/2 0入力ファイルの中身を確認したら、これを specx 実行ファイルに与えて計算を行います。
./specx < in/fe | tee out/fe
< は後ろのファイルを前の実行ファイルに与えるという意味です。
今回の例では in/fe を ./specx に与えています。
単純に ./specx < in/fe とすれば、実行結果が標準出力(要するに画面)に表示されます。
結果をファイルに残しておきたい場合 ./specx < in/fe > out/fe
とすれば良いのですが、今度は画面上に表示されなくなります。
> の代わりに | tee を利用すると、結果を画面に表示しつつファイルにも保存することが可能になります。
以下のような結果が得られていれば成功です。
19-Dec-2020
18:36:03
meshr mse ng mxl
400 35 15 3
data read in
go=go file=data/fe
brvtyp=bcc a= 5.27000 c/a= 0.00000 b/a= 0.00000
alpha= 0.0 beta= 0.0 gamma= 0.0
edelt= 1.0E-03 ewidth= 1.000 reltyp=nrl sdftyp=mjwasa
magtyp=mag record=2nd outtyp=update bzqlty=8
maxitr=100 pmix= 0.03500 mixtyp=tchb-brydn
ntyp= 1 natm= 1 ncmpx= 1
~ (中略) ~
sbtime report
routine 1 2 3 4
count 280 280 280 70
cpu(sec) 16.73 6.80 4.11 0.10
total cpu 10.30 sec
elapsed time 2.83 sec ( 4 threads)
状態密度・バンド構造描画用プログラムのインストール
次に状態密度やバンド構造の計算を行う事を考えます。 既に状態密度やバンド構造を計算することが可能ですが、その前にその計算結果をプロットするための補助プログラムをインストールしておいた方が良いでしょう。 具体的には、以下のように端末に打ち込みます。
make spc gpd状態密度の計算
状態密度の計算を行います。 状態密度やバンド構造の計算を行う場合、その前にgo計算を行っておく必要があります。 状態密度計算用の入力ファイルを作るために、まずgo計算の入力ファイルをコピーし、コピー後のファイルを編集します。
cp in/fe in/fe-dos
gedit in/fe-dos
絶対に必要なのは、入力ファイルの冒頭の go を dos に変更するだけです。
ただ実際には bzqlty の値を大きくした方が見栄えが良くなります。
今回は bzqlty の値を 12 にしてみます。
結果として入力ファイルは以下のようになります。
c----------------------Fe------------------------------------
dos data/fe
c------------------------------------------------------------
c brvtyp a c/a b/a alpha beta gamma
bcc 5.27 , , , , , ,
c------------------------------------------------------------
c edelt ewidth reltyp sdftyp magtyp record
0.001 1.0 nrl mjwasa mag 2nd
c------------------------------------------------------------
c outtyp bzqlty maxitr pmix
update 12 100 0.035
c------------------------------------------------------------
c ntyp
1
c------------------------------------------------------------
c type ncmp rmt field mxl anclr conc
Fe 1 0 0.0 2 26 100
c------------------------------------------------------------
c natm
1
c------------------------------------------------------------
c atmicx(in the unit of a) atmtyp
0 0 0 Fe
c
c------ The following types of inputs and their combination
c are also allowed.
c------ In those cases a, b, b mean primitive unit vectors
c and x, y, z mean conventionnal cell vectors along
c x, y, and z axses. Bare numbers indicate cartesian
c coordinate in the unit of lattice constant a.
c 1/2 1/2 1/2 Fe
c 0a 0b 0c Fe
c 0x 0y 0z Fe
c 1/2a 1/2b 1/2c Fe
c------------------------------------------------------------
c "end" means the end of input data
c "end" is not necessary unless other data used only
c for "spc" exist below.
c----------------------Fe------------------------------------
#end
# The following data are used in the 'spc' mode.
# number of k-point along symmetry line
300
# For bcc bravais lattice, the dispersion relation along
# G-H-N-P-G-N symmetry points (below) is usually calculated.
0 0 0
0 1 0
1/2 1/2 0
1/2 1/2 1/2
0 0 0
1/2 1/2 0
go 計算を行ったときと同様に specx プログラムに入力ファイルを渡します。
./specx < in/fe-dos | tee out/fe-dos
go 計算のときのように、ずらずらと結果が出力されると思います。
この結果を格納した out/fe-dos を状態密度プロット用のプログラムである gpd へ渡します。
./gpd out/fe-dos
この gpd プログラムにファイルを渡すときには、リダイレクトの < を付けてはいけません。
間違って付けると、グラフを描画してくれず Usage: gpd file と怒られます。
私もよく間違えます…
すると以下のようなグラフが描画されるはずです。
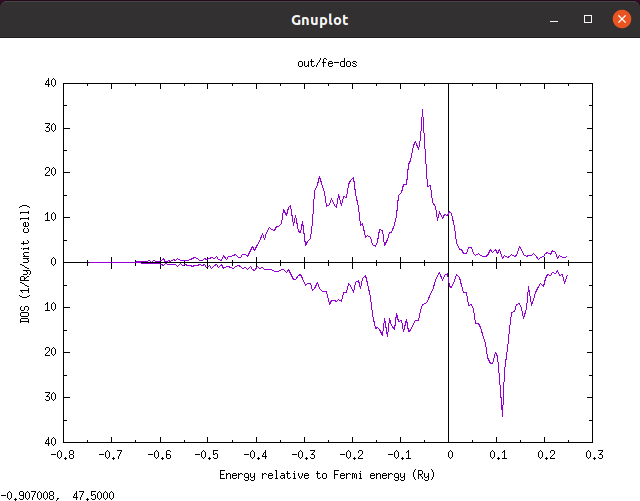
バンド構造の計算
次にバンド構造の計算を行ってみます。
これも状態密度の計算とほぼ同じ手順で、バンド構造計算用の入力ファイルの作成、specx の実行、spc コマンドでの描画、という手続きを踏みます。
cp in/fe in/fe-spc
gedit in/fe-spc
編集個所は go を spc に変更する事、そしてファイルの最後に計算するk点のパスを追加することです。
とは言え、鉄の計算はチュートリアルなのでk点のパスもすでに書き込んであります。
結局、入力ファイルは以下のようになりました。
c----------------------Fe------------------------------------
spc data/fe
c------------------------------------------------------------
c brvtyp a c/a b/a alpha beta gamma
bcc 5.27 , , , , , ,
c------------------------------------------------------------
c edelt ewidth reltyp sdftyp magtyp record
0.001 1.0 nrl mjwasa mag 2nd
c------------------------------------------------------------
c outtyp bzqlty maxitr pmix
update 8 100 0.035
c------------------------------------------------------------
c ntyp
1
c------------------------------------------------------------
c type ncmp rmt field mxl anclr conc
Fe 1 0 0.0 2 26 100
c------------------------------------------------------------
c natm
1
c------------------------------------------------------------
c atmicx(in the unit of a) atmtyp
0 0 0 Fe
c
c------ The following types of inputs and their combination
c are also allowed.
c------ In those cases a, b, b mean primitive unit vectors
c and x, y, z mean conventionnal cell vectors along
c x, y, and z axses. Bare numbers indicate cartesian
c coordinate in the unit of lattice constant a.
c 1/2 1/2 1/2 Fe
c 0a 0b 0c Fe
c 0x 0y 0z Fe
c 1/2a 1/2b 1/2c Fe
c------------------------------------------------------------
c "end" means the end of input data
c "end" is not necessary unless other data used only
c for "spc" exist below.
c----------------------Fe------------------------------------
#end
# The following data are used in the 'spc' mode.
# number of k-point along symmetry line
300
# For bcc bravais lattice, the dispersion relation along
# G-H-N-P-G-N symmetry points (below) is usually calculated.
0 0 0
0 1 0
1/2 1/2 0
1/2 1/2 1/2
0 0 0
1/2 1/2 0
以下のコマンドで specx に入力ファイルを渡します。
./specx < in/fe-spc | tee out/fe-spc
gfortran でコンパイルしている場合、Error termination. Backtrace: とかいうエラーが出てびっくりしますが、実際には正しく計算できています。
また、バンド構造の計算結果は data/fe_up.spc と data/fe_dn.spc に保存され、ターミナルに表示させた情報はあまり有用ではないので out/fe-spc に保存する意味は薄いかもしれません。
なので ./specx < in/fe-spc として出力を捨ててしまってもかまいません。
計算が完了したら、バンド構造描画用プログラム spc に、バンド構造のデータを渡します。
アップスピンのデータは data/fe_up.spc に格納されているので、以下のようにターミナルに打ち込みます。
./spc data/fe_up.spc
すると以下のような画像が表示されるはずです。
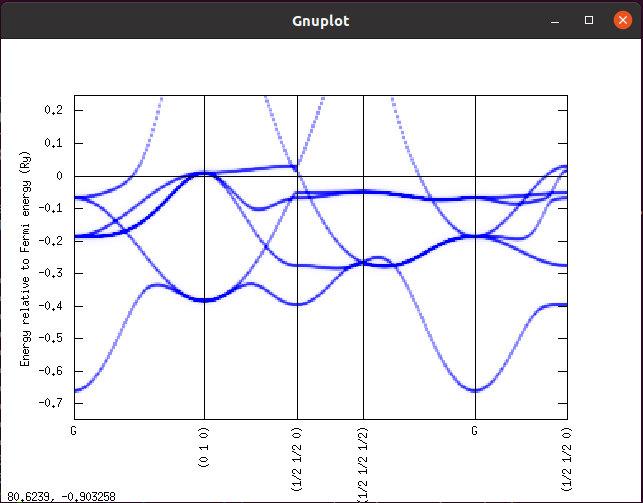
なお、go 計算や dos に関しても当てはまるのですが、AkaiKKR はアップスピンとダウンスピンが逆になっていることがしばしばあります。
同様に ./spc data/fe_dn.spc とするとダウンスピンのバンド構造が描画されます。
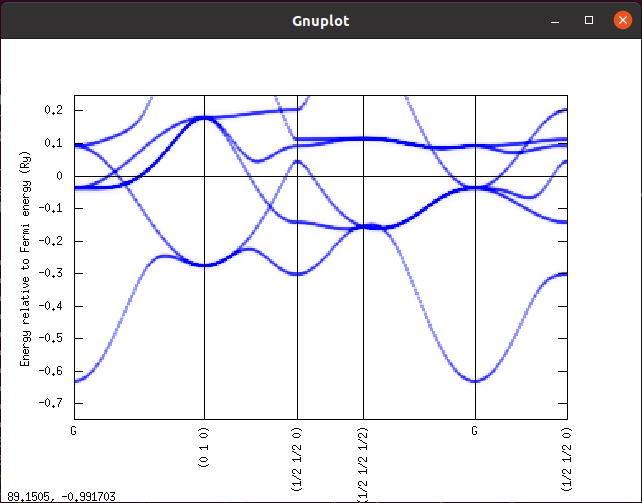
以上で AkaiKKR のインストールとテスト計算は完了です。
パスの設定
ここまでで AkaiKKR が問題なく実行できるようになっています。 しかし、計算プロジェクトごとにディレクトリを作るなどしたときに AkaiKKR の実行ファイルにパスが通っていると便利です。
色々な流儀があると思いますが、ここでは最新版に対してリンクを作っておいて、そのリンクにパスを通すという(ちょっとめんどくさい)事をしています。そんなめんどくさいことしなくてもよかったかも…
まず AkaiKKR の最新版が入っているディレクトリにリンクを作成します。
ln -s ~/kkr/20200910/cpa2002v010/ ~/kkr/cpa2002v010次に ~/.bashrc を編集して、上記のリンクにパスを通します。
gedit ~/.bashrc## *** AkaiKKR ***
export PATH=~/kkr/cpa2002v010:$PATH編集した ~/.bashrc を有効化するのを忘れずに。 以下のコマンドを実行するか、いったんログアウトしてログインしなおせば有効化されます。
source ~/.bashrcアップデート
バージョンごとにディレクトリを作ることにしているので、新しいバージョンに入れ替えたいときにもやることは同じです。 ここでは November 24, 2021 バージョンをインストールし、リンクを作り直す手順を書きます。 やっていることは上と同じなので、特に解説はつけません。 新しいバージョンの AkaiKKR は既にダウンロード済みで ~/Downloads/AkaiKKR/20211124/cpa2002v010.tgz にアーカイブが置いてあるものとします。
mkdir ~/kkr/20211124
cd ~/kkr/20211124
tar xzvf ~/Downloads/AkaiKKR/20211124/cpa2002v010.tgz
cd cpa2002v010/makefile を編集して gfortran でコンパイルする設定にします。
gedit makefile#######################################
# specify the fortran compiler
#fort = ifort
fort = gfortran
#######################################
他にも編集したい部分があれば、それも編集します。
ここでは、一例として source/bzmesh.f の end の前に write(*,'(3x,3(a,i3))')'nfa=',nfa,' nfb=',nfb,' nfc=',nfc を追加します。
gedit source/bzmesh.f 80 continue
deallocate(iwt)
write(*,'(3x,3(a,i3))')'nfa=',nfa,' nfb=',nfb,' nfc=',nfc
endmake
make gpd spc
rm ~/kkr/cpa2002v010
ln -s ~/kkr/20211124/cpa2002v010/ ~/kkr/cpa2002v010フェルミ面の断面描画
バンド構造(ブロッホスペクトル関数)計算の応用でフェルミ面の断面の描画ができます。 このパートは標準的な AkaiKKR のインストール手順からは大きく外れているので、ある程度以上 AkaiKKR の使い方に慣れてからチャレンジするのが良いと思います。
まずは、フェルミ面の断面描画用の実行ファイルを作成する必要があります。 とりあえず、普段使いの specx をバックアップしておきます。
cd ~/kkr/20200910/cpa2002v010/
mv specx specx.back
AkaiKKRの source ディレクトリにある3つのソースコードを編集します。
まずは source/specx.f を開いて67行目ぐらいにある msex の値を 201 から 2 に変更します。
gedit source/specx.f data ef0/7d-1/, dex/5d-2/, emxr/1d0/, msex/2/,mse0/35/
msex は状態密度やバンド構造を計算する際の分解能です。
今回はフェルミ面の断面の描画なので、フェルミ準位近傍の計算だけすればよいという事で2点だけ計算する設定にしてあります。
つぎに source/cemesr.f の17行目ぐらいにある data ref/0.75d0/ を 0.75 から 0.5 に変更します。
gedit source/cemesr.f data ref/0.5d0/このパラメータは、状態密度やバンド構造を計算する際のフェルミ準位の上下のエネルギー範囲の割合を決めています。デフォルトの値である0.75にしておくと、計算するエネルギー範囲に対して4分の3がフェルミ準位の下に、4分の1がフェルミ準位の上になるようになっています。 0.5 にすると計算するエネルギー範囲の真ん中にフェルミ準位が来るようになります。
最後に source/spmain.f の493-494行目ぐらいにある irdfmt と iwrtfmt の値を両方とも 3 から 1 に変更します。
gedit source/spmain.f irdfmt=1
iwrfmt=1
これら二つのパラメータは バンド構造計算(spc計算)の入出力のフォーマットを指定しています。古いバージョンの AkaiKKR では、現在の irdfmt=1, iwrtfmt=1 のフォーマットしかなかったのですが、バンド構造描画プログラム spc が追加されてから、よりバンド構造描画に適した、特徴的なk点のパスを通るような指定がやりやすいフォーマットがデフォルトになりました。
今回は、計算したいk点空間の平面をサンプリングするので、任意のk点を指定できる古いフォーマットの方が適しています。
これら3つのファイルの編集を行ったら make を実行して、新しい実行ファイルを作成します。
make無事 specx が出来たら specxFS (など)に名前を変更して、バックアップしておいた specx を元に戻します。
mv specx specxFS
mv specx.back specxまた編集した3つのソースコードも元に戻しておいた方がいいでしょう。 これでフェルミ面の断面を描画するための AkaiKKR の実行ファイルが出来ました。
次にフェルミ面の断面描画のサンプルを見てみます。 このサンプルは GitLab で公開されている tcsh のシェルスクリプトで、内部で sed と awk を利用しています。 必要なパッケージを導入したのち、スクリプト本体を git を用いてダウンロードしてきます。
sudo apt -y install tcsh sed gawk git
git clone https://gitlab.com/HitoshiGomi/cross-section-of-fermi-surface-for-akaikkr.git
cd cross-section-of-fermi-surface-for-akaikkr/NaWO3/なにはともあれ go 計算を行います。 in/NaWO3.in には go 計算と dos 計算の両方の計算をするように書かれているので状態密度も計算されます。
dos 計算用の bzqlty を少し大きく取りすぎた気もします。 Virtual Box 上で動かしているなど、メモリが少ない環境の場合は bzqlty を下げてもかまいません。
specx < in/NaWO3.in | tee out/NaWO3.out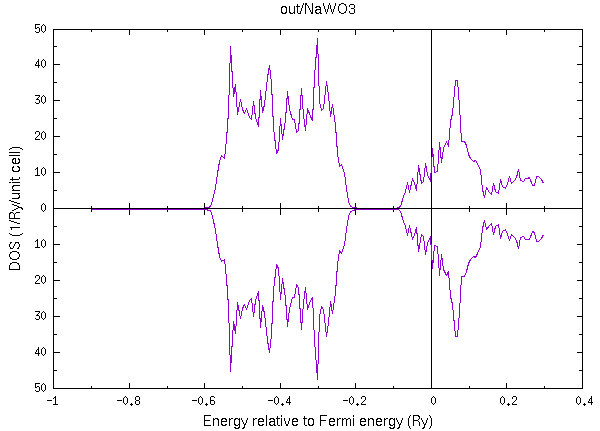
go計算とdos計算が正しくできていれば、上記の様な状態密度が得られるはずです。 ここまではフェルミ面の断面描画とは無関係に実行できるはずの部分です。
次に、このパートで新しくコンパイルした specxFS を試してみます。 入力ファイルは in/NaWO3-Gamma.in です。 この入力ファイルでは、Γ点のブロッホスペクトル関数をフェルミ準位近傍だけで計算します。
specxFS < in/NaWO3-Gamma.in | tee out/NaWO3-Gamma.out計算が正しく終了すると data/NaWO3_up.spc というファイルができているはずです。 このファイルの中身は以下のような感じになっているはずです。
# A(E,k) for spin = 1
0.0000000 0.0010000 0.2749075上記の場合はΓ点だけの計算なので1行しかありません。 ここまでうまくいっていれば、次にシェルスクリプトを実行します。 このシェルスクリプトの実行には、相当な時間がかかります。
chmod +x NaWO3-FS.sh
./NaWO3-FS.shシェルスクリプトを実行すると specxFS が存在しないというエラーが出ることがあるようです。 その場合 NaWO3.sh の中の execfs という変数に specxFS のフルパスを入れてみてください。
gedit NaWO3.sh## Execute file name
#set execfs="specxFS"
set execfs="~/kkr/20200910/cpa2002v010/specxFS"シェルスクリプトが正常に修了したら in/ out/ data/ などのディレクトリに連番のファイルがたくさんできているはずです。 出来たファイルで特に重要なのが analysis/NaWO3-FS_up.dat です。 このファイルをプロットするgnuplotスクリプトが analysis/NaWO3-FS.plt です。 analysis/ に移動して gnuplot に読み込ませます。
cd analysis/
gnuplot -persist NaWO3-FS.pltするとフェルミ面の断面が描画され、PNG形式の画像ファイルが作成されます。
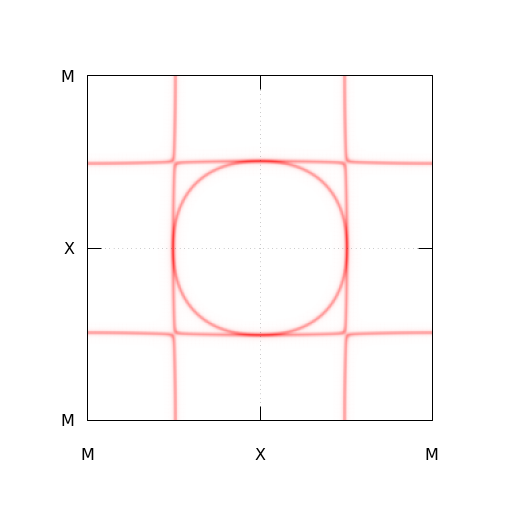
上記のような画像が得られれば成功です。 NaWO3以外の物質のフェルミ面の断面を描画したい場合は NaWO3-FS.sh のコメントを読みながら Please edit FROM here. と Please edit UNTIL here. の間を編集していきます。 チュートリアルで行ったように、まずは普通にgo計算とdos計算を実行し、それらの入力ファイルから in/NaWO3-Gamma.in や template/NaWO3-FS_Template.in に相当するファイルも作る必要があります。